Bio+MedVis Challenge @ IEEE VIS 2022
Taming protein beasts through visualization
How do 3D protein modifications relate to rare disease?
Goal
Understanding chemical modifications on the 3D structure of proteins has the potential to unlock cures for myriad rare diseases. This year, we provide a unique dataset of different types of new protein modifications. We want your help to visually understand where protein modifications are most likely to occur, and whether these are in loci with known connections to specific rare diseases.
Background
Proteins are the workhorses of cells. Their behavior can be regulated by chemical modifications called post-translational modifications (PTMs). PTMs can be identified using technologies such as mass spectrometry (MS). However, proteins can encounter reactive chemicals as well, which occasionally attach themselves to proteins, creating in vivo artifacts. In addition, during analysis, proteins are subject to several processing steps, which introduce further intended and unintended chemical modifications, leading to additional in vitro artifacts. All of these modifications end up as additional layers of “decoration” on the proteins, which are also included in the MS analysis.
Recently, researchers from VIB (Flemish Institute for Biotechnology, Ghent, Belgium) have developed Artificial Intelligence (AI) models [1-3] that can identify PTMs in an unbiased way. Remarkably, they have revealed that proteins can carry many more types of modifications than previously thought, highlighting the need to revise our way of visualizing proteins and their modifications. The formerly relatively simple protein sequences and structures have become “beasts” that we have yet to truly understand! Can you tame the discovered protein “beasts” with the use of insightful, inspiring, and interactive visuals? Challenge accepted!
Data Set
The data are provided by domain experts from the CompOmics group at VIB and Ghent University in Ghent, Belgium. These include datasets for three proteins known to be important in rare diseases [4]: i) Aldolase A (ALDOA) protein in Hereditary nonspherocytic hemolytic anemia (HNSHA); ii) Heterogeneous nuclear ribonucleoprotein A1 (HNRNPA1) protein in Amyotrophic lateral sclerosis; and iii) Transforming growth factor beta 1 (TGFB1) protein in Camurati–Engelmann disease.
For each protein, we provide i) a tab-delimited file that lists its modified loci (residues) and the modification type. These modifications were identified across human proteins in the publicly available proteomics identification database PRIDE [5] using the machine-learning based ionbot search engine [3]. Notably, a single residue may carry multiple types of modifications, but only one of these at a time; ii) annotations of known mutations that alter protein function, iii) structure files for these proteins (either experimental structures, or AlphaFold [6] predictions).
Download link: 2022_BioMedVisChallenge_Data_Ver2.zip
(2022-08-22: Updated data set with the protein ATP-dependent RNA helicase DDX3X that can be used for the re-design
challenge.)
Tasks
Once you relate the detected modifications to the protein 3D structure (for instance, unstructured regions are more likely hot-spots for modifications), the first task is to visualize multiple, potentially overlapping modifications on a protein structure in a user-intuitive way.
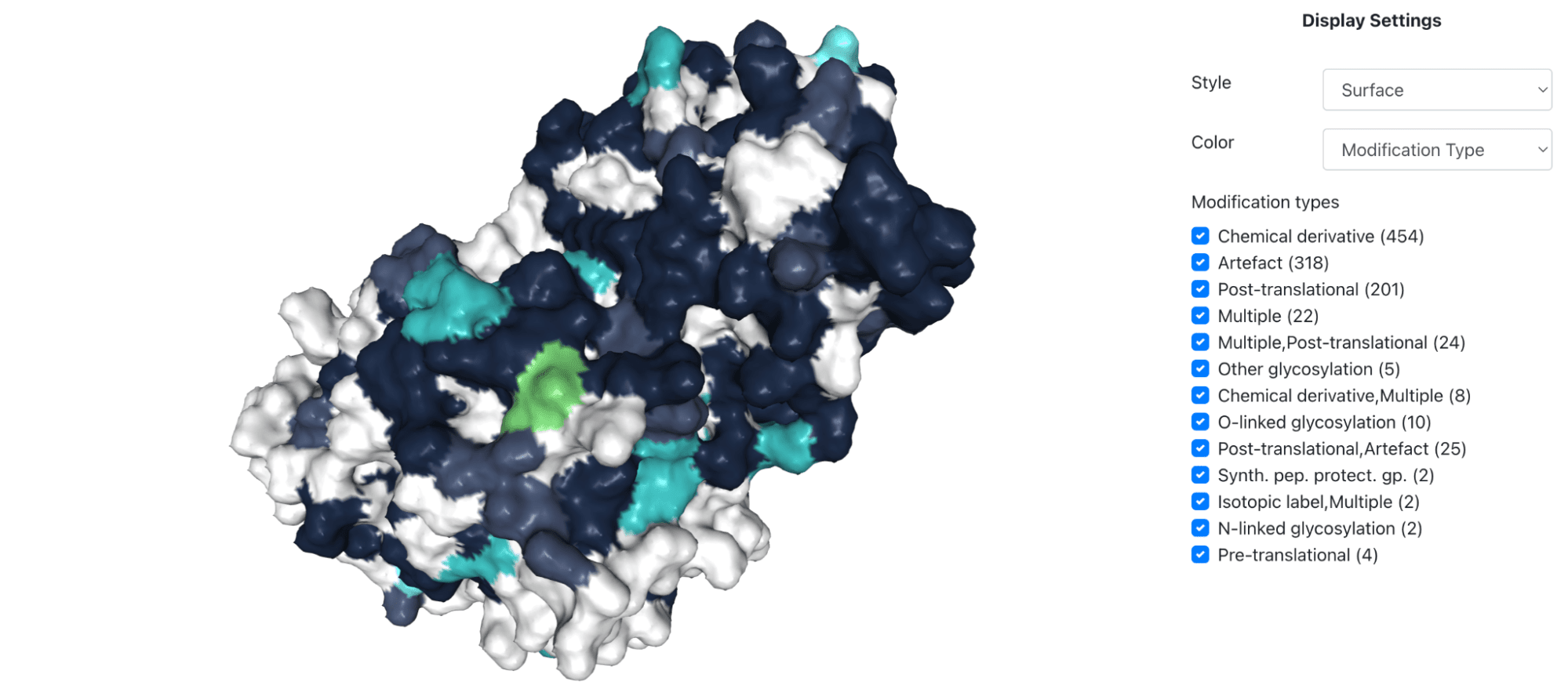
The second task is to relate a protein mutation that is known to be involved in a specific rare disease with its modification status, as well as those of residues proximal to it (proximity either by sequence or structure). This is because a mutation can alter protein function by a variety of mechanisms, one of which is the alteration of a (proximal) modification site. Please note that this is because a mutation can render some of these modifications of the protein chemically impossible. For this challenge task, we provide you with several important mutations known to be involved in the proteins and rare diseases (i-iii) listed above.
Complementary challenge: Re-design an existing visualization
If you do not want to deep dive into the tasks above, you can participate in an alternative challenge! This re-design challenge is to take an existing visualization as a starting point. Your task is to revise and improve the visualization in the figures below based on fundamental visualization principles. These visualizations show all PTMs identified on protein ATP-dependent RNA helicase DDX3X; these PTMs can alter the enzyme activity, thus affecting protein translation and cell growth. Due to the large number of modifications and modified residues, visualizations can become extremely cluttered (see Figure 3), which is the main problem to address in this redesign challenge. An important aspect to emphasize is how multiple modifications can occur on the same site, as this implies substrate competition between different protein-modifying enzymes.
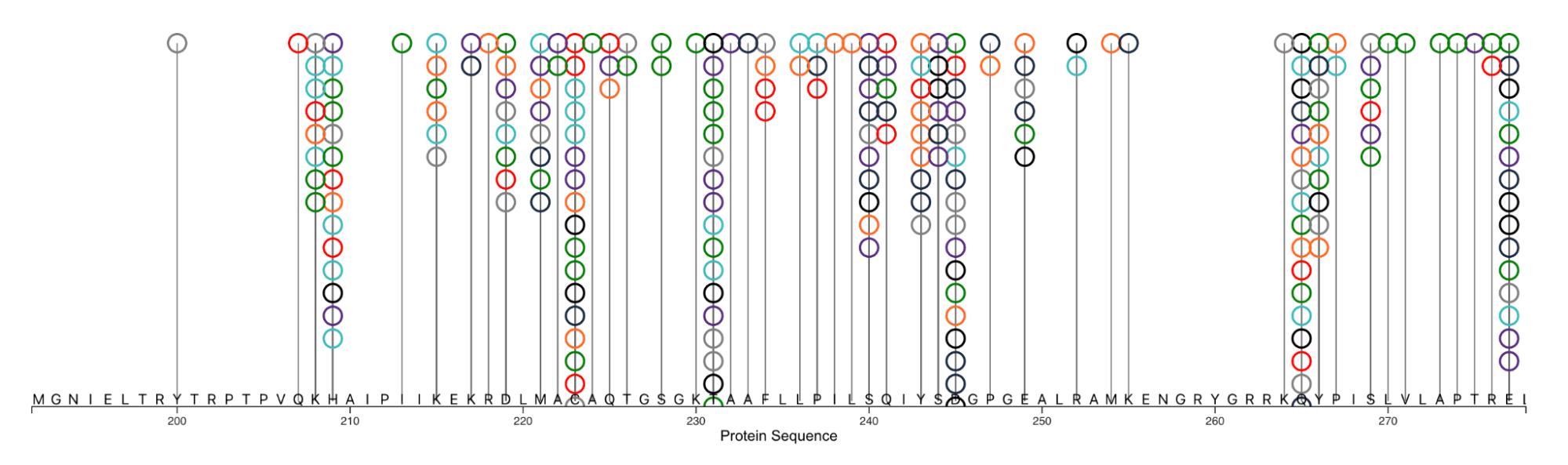
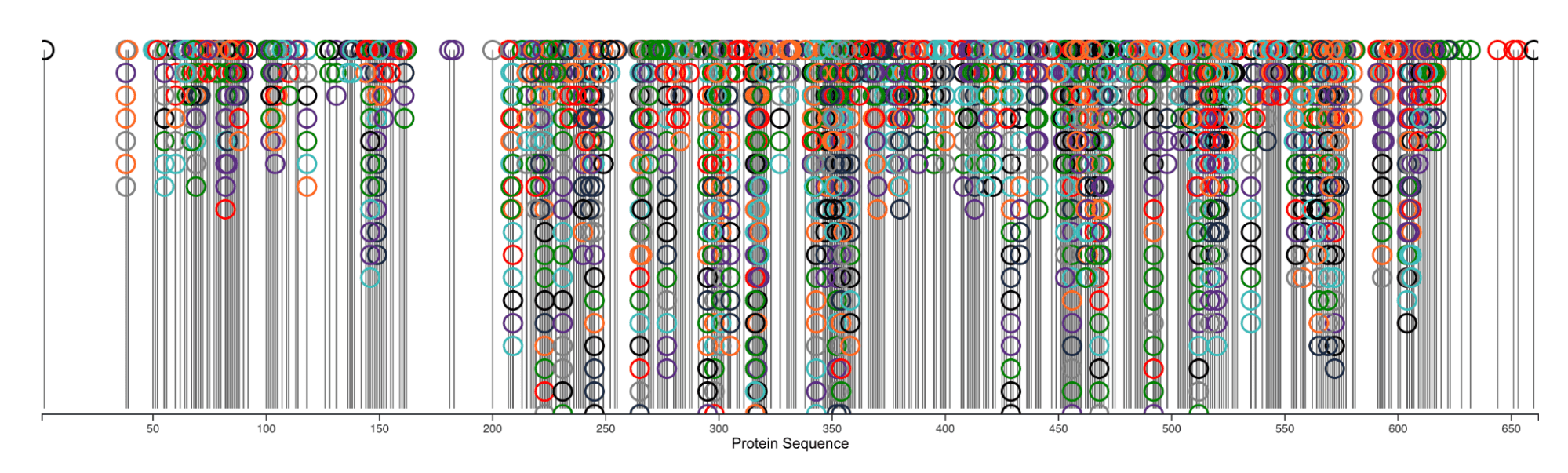
Submission
Submissions will be considered for talk or poster presentations. Please send a two-page PDF abstract with unlimited additional figures and a draft of your proposed poster (max 10MB) to biovis_challenge@ieeevis.org latest on September 9th 2022 (anywhere on earth). The abstract should include:
- aspects of the figure identified as needing improvement or clarification,
- justification of encoding and design choices,
- at least one or more images of your design
- optional: a video or screencast to explain the visual encoding
Last year, the acceptance rate for presentations happened to be 100%, yet such acceptance rate is not guaranteed. Selected submissions will be invited for talk presentations during the Bio+MedVis session within the IEEE VIS 2022 conference.
Questions?
Please feel free to ask questions on our Slack channel if you need more
information and details about the data or tasks:
biovis2016.slack.com » #biovis-challenge2022
Otherwise, feel free to contact us at: biovis_challenge@ieeevis.org
The chairs of the Bio+MedVis Challenge @ IEEE VIS:
- Zeynep Gumus (Icahn School of Medicine at Mount Sinai, USA) [zeynep.gumus (at) mssm.edu]
- Thomas Höllt (TU Delft, Netherlands) [T.Hollt-1 (at) tudelft.nl]
- Daniel Jönsson (Linköping University, Sweden) [daniel.jonsson (at) liu.se]
- Renata Raidou (TU Wien, Austria) [rraidou (at) cg.tuwien.ac.at]
References
[1] Gabriels, R., Martens, L. and Degroeve, S., 2019. Updated MS²PIP web server delivers fast and accurate MS² peak intensity prediction for multiple fragmentation methods, instruments and labeling techniques. Nucleic acids research, 47(W1), pp.W295-W299. https://pubmed.ncbi.nlm.nih.gov/31028400/
[2] Bouwmeester, R., Gabriels, R., Hulstaert, N., Martens, L. and Degroeve, S., 2021. DeepLC can predict retention times for peptides that carry as-yet unseen modifications. Nature methods, 18(11), pp.1363-1369. https://pubmed.ncbi.nlm.nih.gov/34711972/
[3] Degroeve, S., Gabriels, R., Velghe, K., Bouwmeester, R., Tichshenko, N. and Martens, L., 2021. ionbot: a novel, innovative and sensitive machine learning approach to LC-MS/MS peptide identification. https://www.biorxiv.org/content/10.1101/2021.07.02.450686v2
[4] https://www.ncbi.nlm.nih.gov/clinvar/
[5] Martens, L., Hermjakob, H., Jones, P., Adamski, M., Taylor, C., States, D., Gevaert, K., Vandekerckhove, J. and Apweiler, R., 2005. PRIDE: the proteomics identifications database. Proteomics, 5(13), pp.3537-3545. https://pubmed.ncbi.nlm.nih.gov/16041671/