BioVis 2011 Paper
MDMap : A System for Data-driven Layout and Exploration of Molecular Dynamics Simulations
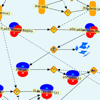
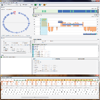
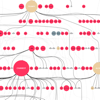
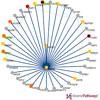
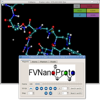
Contemporary molecular dynamics simulations result in a glut of simulation data, making analysis and discovery a difficult and burdensome task. We present MDMap, a system designed to summarize long-running molecular dynamics (MD) simulations. We represent a molecular dynamics simulation as a state transition graph over a set of intermediate (stable and semi-stable) states. The transitions amongst the states together with their frequencies represent the flow of a biomolecule through the trajectory space. MDMap automatically determines potential intermediate conformations and the transitions amongst them by analyzing the conformational space explored by the MD simulation. MDMap is an automated system to visualize MD simulations as state-transition diagrams, and can replace the current tedious manual layouts of biomolecular folding landscapes with an automated tool. The layout of the representative states and the corresponding transitions among them is presented to the user as a visual synopsis of the long-running MD simulation. We compare and contrast multiple presentations of the state transition diagrams, such as conformational embedding, and spectral, hierarchical, and force-directed graph layouts. We believe this system could provide a road-map for the visualization of other stochastic time-varying simulations in a variety of different domains.
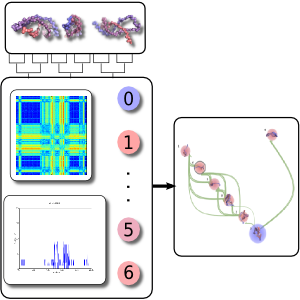
BioVis 2011 Papers and Abstracts