BioVis 2011 Paper
Parallel Contour-Buildup Algorithm for the Molecular Surface
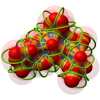
Molecular Dynamics simulations are an essential tool for many applications. The simulation of large molecules-like proteins-over long trajectories is of high importance e. g. for pharmaceutical, biochemical and medical research. For analyzing these data sets interactive visualization plays a crucial role as details of the interactions of molecules are often affected by the spatial relations between these molecules. From the large range of visual representations for such data, molecule surface representations are of high importance as they clearly depict geometric interactions, such as docking or substrate channel accessibility. However, these surface visualizations are computationally demanding and thus pose a challenge for interactive visualization of time-dependent data sets. We propose an optimization of the Contour-Buildup algorithm for the Solvent Excluded Surface (SES) to remedy this issue. An optimized subdivision of calculation tasks of the original algorithm allows for full utilization of massive parallel processing hardware. Our approach is especially well suited for modern graphics hardware employing the CUDA programming language. As we do not rely on any pre-computations our method is intrinsically applicable to time-dependent data with arbitrarily long trajectories. We are able to visualize the SES for molecules with up to ten thousand atoms interactively on standard consumer graphics cards.
BioVis 2011 Papers and Abstracts